Potential-Bound and Rigidly
Bound Rotor Energy Distribution in 2 Dimensions
Introduction
We have
previously shown animations of rigid rotors where the separation between the
end atoms is constant. Here we will
discuss the energy distributions attained by rotors that are bound by a strong
potential energy variation. This will be
a more realistic model of diatomic molecule
motion and energy distributions.
For this model, all of the forces between particles will be mediated by
central forces similar to the those of the Lennard-Jones potential. Central force here means that the force is
along the line separating the interacting particles and the value of the force
is a function of the separation. For
microscopic particles there are very few exceptions to this type of force.
An
important revelation that we come to as a result of this simulation is that some of the terms
used in discussing energy distributions of complex particles are misleading. In particular the term "degrees of
freedom" as applied to the energy distributions and heat capacity needs to
be changed to "modes of energy interchange". See the section below for a discussion of
this.
Another
revelation is that two revered tenets of physics, linear and angular momentum
conservation, are never used in this simulation (in fact it appears that these
conservation principles are really only a mathematical convenience, mostly
useful for hard body interactions). Momentum conservation needs to be renamed to
"directed energy conservation".
See the section below for a discussion of this.
Last but
not least, I have included a section on how I set up the initial parameters of
the rotors according to their rotational speed.
Energy Distribution Functional Results
Simple 2
dimensional constrained atoms have the energy distribution:
|
|
|
(1.1)
|
where f(E) is normalized to a peak value of 1.0.
For rotors bound by a Lennard-Jones type potential and
constrained to 2 linear dimensions we obtain the following distribution also
normalized to 1 at its peak:
|
|
|
(1.2)
|
Figure 1 shows the resulting plots for the potential-bound
of rotor as well as the plot for atoms in equilibrium with the rotors.
For rigidly bound rotors and constrained to 2 linear
dimensions we obtain the following distribution also normalized to 1 at its
peak:
|
|
|
(1.3)
|
Figure 2 shows the resulting plots for the rigid rotor as
well as the plot for atoms in equilibrium with the rotors.

Figure 1.Distribution
of Energies of both Atoms and Potential Bound Rotors.
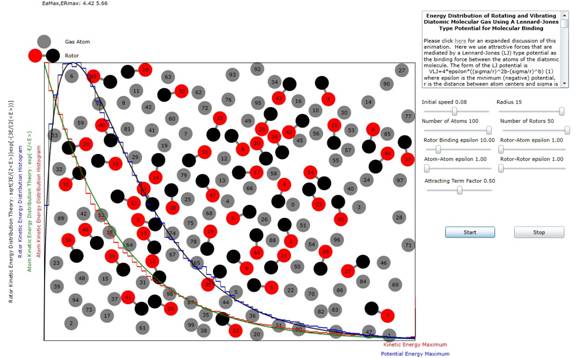
Figure 2. Distribution
of Energies of both Atoms and Rigidly Bound Rotors.
Renaming the Term "Degrees of Freedom" in Reference to
Statistical Energy Distributions
An
important revelation that we come to as a result of this simulation is that some of the terms
used in discussing energy distributions of complex particles are misleading. In particular the term "degrees of
freedom" as applied to the energy distributions and heat capacity needs to
be changed to "modes of energy
interchange". The reason for
this change is that we have here achieved what would normally be called an
increase of the degrees of freedom, not by increasing
freedom of movement, but by applying
a variable constraint. For example,
a gas of particles that can move in two dimensions has two modes of energy
interchange, (x1,y1) may become (x2,y2) and vice-versa. A gas of particles that can move in only one
direction has only one mode of energy interchange, x1 may become x2. For our non-rigid rotors, energy interchange
can occur 4 ways. Two of these modes are the simple linear interchange of the
center of mass motions mentioned above.
Another is the conversion of center of mass energy to rotational energy
and vice-versa. Yet another is the
conversion of center of mass energy to rotational energy and vice-versa. Since vibrational motion is orthogonal to
rotational motion for a diatomic atom, there is no direct way for energy
conversion between these two motions to happen.
Renaming Linear and Angular Momentum Conservation
An
important revelation of this simulation is that two venerable tenets of
physics, linear and angular momentum conservation, are never used in this
simulation. In fact, it appears that these conservation principles are really
only a mathematical convenience, mostly useful for hard body interactions. The only way that either of these properties
change is via an interaction with a spatially and time varying potential
energy. This potential energy can
either yield kinetic energy or accept kinetic energy of the particles by
changing their velocity.
It would at
first appear that the particle's change of velocity could be in any direction
and that would still satisfy the requirement of energy conservation. However, one must take into account that an
arbitrary direction of the velocity change has a very important impact, through
time integration of this velocity change, on the new positions of the
particle. If that position turns out to
be not in agreement with the force provided by the potential, then energy on
subsequent integrations of the velocity will not be conserved. So, for interactions between particles which
result in velocity change, I prefer to change the term "momentum
conservation" to a more accurate term "directed energy conservation" which preserves the innate vector nature of energy conservation as
well as the scalar nature of energy conservation.
Directed Energy Conservation
A perfectly
good equation for iteration of velocities and positions is:
|
|
|
(1.4)
|
where the upside down triangle stands for spatial gradient,
T is the kinetic energy and V is the potential energy. Forces that can affect the particle's
velocity are proportional to the gradient of the potential energy along the
direction to the particle.
|
|
|
(1.5)
|
where the carat on top of r stands for unit vector along that direction.
T has the following form
|
|
|
(1.6)
|
where vr is the velocity along the direction r.
Then the gradient of T along the direction of the force is
|
|
|
(1.7)
|
We can use the chain rule to convert this expression to
derivatives with respect to time:
|
|
|
(1.8)
|
The particle's velocity component in the r direction is
updated incrementally using equation (1.8). The position of the particle is then updated
using this new velocity.
Rotor Bound by Lennard-Jones type potential
Rather than making the spacing between the disks rigid, we
can use a potential to keep the rotor disks together. For this we will use a modified Lennard Jones
potential..
The normal LJ potential is given by
|
|
|
(1.9)
|
where the minimum potential is -ε
and σ is usually chosen as a nominal separation
between disks.
We will change this expression by assigning variables to the
power of he σ/r ratios.
|
|
|
|
|
|
|
(1.10)
|
The value of r at the minimum potential is computed by
setting the derivative of V(r) equal to zero and is
|
|
|
(1.11)
|
This is also the separation r where the force between
particles goes to zero. We can use totally similar math to compute the spacing
where maximum attractive force occurs.
First defining a power ratio, Pr, using the second derivative of the
potential we have:
|
|
|
(1.12)
|
we find that the spacing where maximum attractive force
occurs is:
|
|
|
(1.13)
|
and this leads to the value of the maximum attractive force:
|
|
|
(1.14)
|
Using Fmax we can compute the maximum rotational
speed that the diatomic molecule can have, and remain bound, for a given value
of ε:
|
|
|
(1.15)
|
or
|
|
|
(1.16)
|
where m is the mass of a single atom.
We can choose an initial separation for the atoms such that
their separation stays constant by solving for r0 in the following
equation:
|
|
|
(1.17)
|
Equation (1.17) can always be solved
by a Newton-Raphson numerical method or it can be solved algebraically for the
special case a=2b:
For that special case, if we let then we can rewrite equation (1.17)
|
|
|
(1.18)
|
and equation (1.18)
is recognized as a simple quadratic equation.
The solution for u is
|
|
|
(1.19)
|
Again, for the special case a=2b we can write a better
expression for vmax
|
|
|
(1.20)
|
When the rotor is hit by another particle, it could easily
be unbound unless the value of ε for the rotor binding is much larger
than the value for the other particle.
It seems prudent to choose the interaction potential for this kind of
collision to be:
|
|
|
(1.21)
|
where εA
is the value of ε for the atom and εR
is the value for the rotor's binding.
This should tend to maintain the binding of the rotor components.